


Подкаст-версия этой статьи уже на нашем Dental Portal Радио в Telegram. 👉 Слушать подкаст
Содержание
- Что считается медицинским изделием — и почему это важно знать клинике
- Классы риска МИ: от скальпеля до импланта
- Кто обязан регистрировать МИ и кто несёт ответственность
- Как устроена регистрация: этапы, схемы, новые правила 2025 года
- Регистрационное досье: что входит в пакет документов
- Сроки и стоимость регистрации: актуальные данные
- Одноэтапная регистрация для отечественных производителей: новая возможность
- Типичные ошибки, из-за которых получают отказ
- Что меняет Постановление №1684: главное за 5 минут
- Регистрация в ЕАЭС vs национальная регистрация в РФ
- Как владельцу клиники проверить РУ поставщика
- FAQ: ответы на частые вопросы
Что считается медицинским изделием — и почему это важно знать клинике
Медицинское изделие (МИ) — это любой инструмент, аппарат, прибор, оборудование, материал или программное обеспечение, предназначенное для диагностики, профилактики, лечения, мониторинга состояния здоровья или восстановления функций организма. Ключевой критерий: изделие достигает своей цели не через фармакологическое или метаболическое воздействие — иначе это уже лекарство.
В стоматологии к медицинским изделиям относятся буквально сотни позиций: от стоматологического кресла и автоклава до боров, слепочных материалов, имплантов и интраоральных сканеров. Даже программное обеспечение для диагностики кариеса — это МИ, которое нужно регистрировать.
Это нужно понимать каждому участнику рынка. Владелец клиники обязан проверить наличие РУ перед закупкой. Поставщик — иметь действующее РУ на каждую позицию ассортимента. Производитель — пройти регистрацию до вывода продукта на рынок.
Классы риска МИ: от скальпеля до импланта
Почему класс риска определяет всё
Класс потенциального риска — это оценка того, насколько опасным может быть изделие для пациента при неправильном применении или отказе. Именно класс риска определяет объём доказательной документации, необходимость клинических испытаний и итоговые сроки регистрации.
В России выделяют четыре класса риска МИ по Постановлению Правительства РФ №1416:

Кто обязан регистрировать МИ и кто несёт ответственность
Регистрировать МИ обязан заявитель — это юридическое лицо, зарегистрированное в России (или в стране ЕАЭС при регистрации по единым правилам). Иностранный производитель не может самостоятельно подать заявление в Росздравнадзор — ему нужен уполномоченный представитель на территории РФ.
Кто является заявителем на практике
- Российский производитель — подаёт сам от своего имени
- Официальный дистрибьютор иностранного бренда — получает РУ как уполномоченный представитель
- Импортёр — несёт полную ответственность за соответствие продукта требованиям
Ответственность серьёзная. За обращение незарегистрированных МИ предусмотрены административные штрафы по ст. 6.28 КоАП РФ: для должностных лиц — до 30 000 рублей, для юридических — до 1 000 000 рублей, а в ряде случаев — приостановление деятельности.
Владелец клиники также несёт ответственность, если использует незарегистрированные изделия: при проверке Росздравнадзора это будет грубым нарушением лицензионных требований.
Как устроена регистрация: этапы, схемы, новые правила 2026 года
Две схемы регистрации по новым правилам
С 1 марта 2025 года в России действует Постановление Правительства №1684 от 30.11.2024 — новые Правила государственной регистрации медицинских изделий. Теперь существуют две основные схемы:
Схема 1 — Стандартная (для всех производителей)
Применяется по умолчанию. Предусматривает двухэтапную экспертизу: сначала техническая документация, затем — результаты испытаний.
Схема 2 — Одноэтапная (упрощённая для отечественных производителей)
Применяется, если заявитель и производственная площадка находятся в России, а испытания проведены во ВНИИ имитационных медицинских технологий (ВНИИИМТ) и аккредитованных федеральных НМИЦ Минздрава.
Основные этапы регистрации
- Определение класса риска — выбор кода номенклатурной классификации МИ на сайте Росздравнадзора
- Формирование регистрационного досье — сбор технической документации, инструкций, данных испытаний
- Проведение технических и токсикологических испытаний — во ВНИИИМТ или аккредитованных лабораториях
- Клинические испытания (при необходимости — для классов 2б и 3)
- Подача заявления через АИС Росздравнадзора — только в электронном формате, бумажные заявления больше не принимаются
- Экспертиза документации Росздравнадзором
- Получение регистрационного удостоверения — электронное РУ, бумажный вариант упразднён
Все данные о сроках и порядке основаны на анализе нормативных документов и информации из открытых профессиональных источников (Perplexity, топ-30 изданий ниши), а также экспертных оценок специалистов отрасли.
Регистрационное досье: что входит в пакет документов
Регистрационное досье — это полный пакет документов, который подтверждает безопасность и эффективность медицинского изделия. Его содержание зависит от класса риска, но базовая структура единая.
Стандартный состав досье
- Заявление по установленной форме (подаётся через АИС Росздравнадзора)
- Технические условия (ТУ) или стандарт организации (СТО) — описание изделия и требования к нему
- Инструкция по применению — на русском языке, утверждённая производителем
- Макет маркировки — этикетка с обязательными реквизитами
- Протоколы технических испытаний — подтверждают соответствие стандартам
- Протоколы токсикологических испытаний — для изделий, контактирующих с тканями
- Протоколы клинических испытаний — для классов 2б и 3, а также при наличии новых клинических показаний
- Сведения о производителе — юридические документы, данные о системе менеджмента качества (ISO 13485 или ГОСТ Р ИСО 13485)
- Для иностранных производителей — документы уполномоченного представителя и нотариально заверенные переводы
Требования к электронному досье
Согласно новым правилам, все документы подаются в формате PDF/A, с поисковым слоем (OCR) и разрешением не менее 300 dpi. Ни один документ не принимается в виде скана без текстового слоя.
Сроки и стоимость регистрации: актуальные данные
Сроки существенно сократились после принятия ПП №1684.
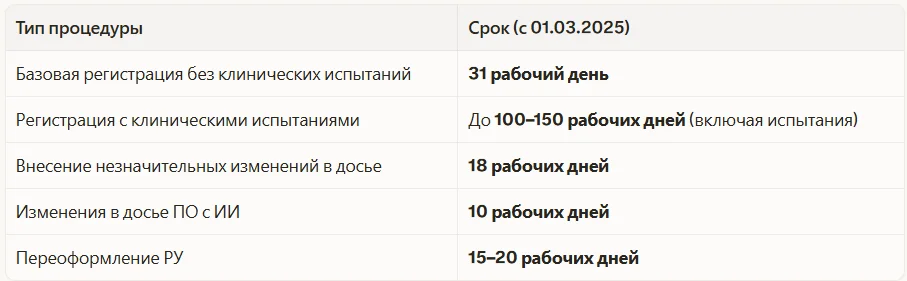
Государственная пошлина за регистрацию МИ в 2025–2026 году составляет 3 000 рублей (за само РУ). Реальные расходы на регистрацию значительно выше — их формируют испытания.
Ориентировочная стоимость полного цикла регистрации
- Технические испытания (ВНИИИМТ или аккредитованная лаборатория): от 100 000 до 500 000 рублей в зависимости от сложности изделия
- Токсикологические испытания: от 50 000 до 300 000 рублей
- Клинические испытания (для 2б и 3 класса): от 1 000 000 рублей и выше
- Услуги регистрационного центра «под ключ»: от 300 000 до 2 000 000 рублей
Данные ориентировочные; точную стоимость запрашивайте у аккредитованных организаций — ВНИИ МТ, регистрационных центров.
Одноэтапная регистрация для отечественных производителей: новая возможность
Это одно из ключевых нововведений ПП №1684 — механизм, который реально ускоряет выход российских МИ на рынок.
Условия для использования одноэтапной схемы:
- Заявитель и производственная площадка зарегистрированы на территории РФ
- Технические и токсикологические испытания проведены во ВНИИ имитационных медицинских технологий (ВНИИ МТ) Росздравнадзора
- Клинические испытания (если необходимы) выполнены в аккредитованных федеральных НМИЦ Минздрава России
При соблюдении этих условий документация рассматривается единожды — повторная экспертиза не проводится. Это экономит реально 3–6 месяцев по сравнению со стандартной процедурой.
Типичные ошибки, из-за которых получают отказ
Анализ практики регистраций показывает несколько устойчивых паттернов ошибок.
ТОП-7 причин отказа или возврата досье
- Неправильно определён класс риска — производитель занижает класс, чтобы избежать клинических испытаний; Росздравнадзор это выявляет
- Документы не соответствуют формату PDF/A — скан без текстового слоя, низкое разрешение, неверная структура файлов
- Инструкция по применению не переведена на русский язык или перевод выполнен некачественно
- Протоколы испытаний устарели — использованы результаты испытаний, полученные более 3 лет назад
- Отсутствие или несоответствие системы менеджмента качества — документы СМК не обновлены под ГОСТ Р ИСО 13485-2017
- Ошибки в маркировке — отсутствуют обязательные реквизиты по ГОСТ Р 51935 или требованиям ЕАЭС
- Несоответствие заявленных характеристик данным испытаний — технические условия обещают одно, а протоколы показывают другое
Что меняет Постановление №1684: главное за 5 минут
ПП РФ №1684 от 30.11.2024 — самое значимое изменение в регулировании МИ за последние годы. Вот что изменилось принципиально:
- Полный переход в электронный формат: бумажное РУ больше не выдаётся — только электронное свидетельство в реестре Росздравнадзора
- Одноэтапная схема для российских производителей при испытаниях в ВНИИ МТ
- Сокращение сроков: базовая регистрация — 31 рабочий день вместо прежних 60+
- Отдельные правила для ПО с ИИ (искусственным интеллектом): выделена в отдельную категорию с 10-дневным сроком для изменений
- Механизм обжалования внутри личного кабинета АИС
- Гармонизация с правилами ЕАЭС — упрощает последующий выход на рынки Беларуси, Казахстана, Армении, Кыргызстана
- Расширение понятийного аппарата — добавлены определения для новых типов изделий, включая цифровые и комбинированные продукты
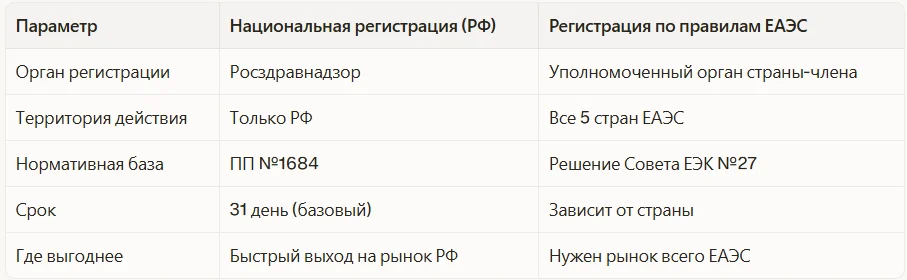
Регистрация в ЕАЭС vs национальная регистрация в РФ
Россия входит в Евразийский экономический союз (ЕАЭС), который формирует единый рынок МИ для пяти стран. Производитель может выбрать путь регистрации.
Как владельцу клиники проверить РУ поставщика
Это практический навык, который защищает клинику от штрафов и репутационных рисков. Проверка занимает буквально 2 минуты.
Пошаговая инструкция проверки РУ
- Зайдите на сайт Росздравнадзора → раздел «Государственный реестр медицинских изделий»
- Введите номер РУ (формат: ФСЗ ГГГГ/NNNNN или РЗН ГГГГ/NNNNN) или название изделия
- Убедитесь, что статус РУ — «Действующее»
- Проверьте, что наименование изделия и производитель совпадают с тем, что написано на упаковке
- Проверьте срок действия — у некоторых старых РУ есть ограниченный срок
Что делать, если РУ не найдено или просрочено:
- Немедленно запросить у поставщика актуальную копию РУ
- Приостановить использование изделия до подтверждения регистрации
- При необходимости проконсультироваться с юристом перед следующей закупкой
Проверяйте РУ при каждой новой позиции в ассортименте поставщика, а не только при первом знакомстве — статус изделия может меняться.
FAQ: ответы на частые вопросы
Нужно ли регистрировать медицинское изделие, если оно уже зарегистрировано в Европе (CE-маркировка)?
Да, обязательно. CE-маркировка не является основанием для допуска к обращению в России. Нужна отдельная регистрация в Росздравнадзоре по российским или ЕАЭС-правилам. Единственное исключение — временные разрешения для отдельных категорий в рамках параллельного импорта, но это разовый механизм с ограниченным сроком действия.
Сколько действует регистрационное удостоверение?
По правилам ПП №1684, РУ выдаётся бессрочно. Но если в конструкцию или состав изделия вносятся изменения, требуется переоформление. Старые РУ с ограниченным сроком (до 2025 года) продлеваются в штатном порядке.
Может ли клиника использовать изделие без РУ в экстренных случаях?
Только в рамках официально разрешённых программ — например, по разрешению Росздравнадзора для конкретного пациента по жизненным показаниям. В обычной практике — нет. Это административное нарушение.
Как зарегистрировать зарубежный имплант, который только вышел на рынок?
Иностранный производитель должен назначить уполномоченного представителя в России — российское юридическое лицо. Именно оно будет заявителем. Без уполномоченного представителя регистрация невозможна.
Нужно ли регистрировать 3D-печатные конструкции, изготовленные в лаборатории?
Это зависит от назначения. Если конструкция изготавливается индивидуально под конкретного пациента и не выводится на рынок как серийное изделие, то регистрация не требуется, но возникают другие требования, такие как использование полимеров имеющих РУ (для хирургических шаблонов). Серийные изделия, в том числе производимые на 3D-принтере, подлежат регистрации в случаях предусмотренных действующим законодательством.
Что будет, если срок действия РУ истёк, но изделие ещё на складе?
Обращение изделия без действующего РУ незаконно — даже если оно было законно куплено до истечения срока. Необходимо либо своевременно переоформить РУ, либо прекратить реализацию.
Изменилось ли что-то для параллельного импорта в 2025 году?
Параллельный импорт МИ в России разрешён как временная мера для компенсации ушедших брендов. Однако требования к безопасности сохраняются: изделие всё равно должно иметь действующее РУ или проходить упрощённую процедуру. Механизм регулярно обновляется Минпромторгом — следите за актуальными перечнями.
Вывод
Регистрация медицинских изделий в России — это не бюрократическая формальность, а гарантия безопасности для пациентов и правовая защита для всех участников рынка. После принятия ПП №1684 процедура стала значительно быстрее и прозрачнее. Производители и дистрибьюторы получили чёткие правила игры и новые инструменты, а владельцы клиник — простой способ проверить законность используемого оборудования и материалов.


